
欧洲医疗器械法规(MDR)的8个问题已回答
在欧洲医疗器械法规(MDR)是一组新的法规管辖在欧洲生产和医疗器械的分布,以及遵守法规是强制性的医疗设备公司希望出售他们的产品在欧洲市场。
如果您的公司已经符合医疗器械指令(MDD),请不要自满 - MDR代表了全新的法规,并带来了很多变化。
作为医疗器械公司的起点,我们创建了这篇文章,回答了关于新MDR的七个最重要的问题。
我们将介绍旧法规需要更新的原因,新MDR文档的结构以及医疗设备公司需要了解的一些最新要求。
1.为什么欧洲医疗器械法规(MDR)需要更新?
MDD需要更新的原因有很多。例如,当MDD于1992年成为法律时,作为医疗设备的软件尚不存在。软件是控制电机的东西,没有患者可以用来监控自己健康的应用程序。
自1992年以来,欧洲的人口统计数据也发生了变化,人口越来越多,人们越来越倾向于向公众提供医疗设备技术信息的透明度。
这些因素加在一起的原因是MDD被一项新指令所取代,该指令将鼓励更广泛地遵守整个欧洲的标准医疗设备法规。
2.新欧洲医疗器械法规(MDR)如何构建?
新的MDR文件长度为174页。它包含13页的介绍,随后是10章(79页)中的123篇文章和17个附件(80页)。与长度为60页的MDD相比,新法规更长,更详细。除此之外,还发布了42项实施法案,用于澄清/实施MDR和12项授权法案,用于修改/修改。
这个新内容的原因是什么?您可能不知道有源植入式医疗器械指令(AIMD)也已被整合到新的MDR中,但除此之外,还有很多以前不存在的内容。
3. 欧洲医疗器械法规(MDR)的主要主题变化是什么?
与其前身MDD相比,新的欧洲MDR不太关注医疗器械制造的预批准阶段,而是推动医疗器械监管的生命周期方法。
虽然旧的MDD基本上是医疗器械公司如何获得CE标志并进入市场的手册,但新法规鼓励在整个产品生命周期中提升医疗器械公司产品责任的政策和程序。
欧洲市场包括27个成员国(不包括英国)以及落入欧洲经济区(列支敦士登,挪威,冰岛)的其他成员国。该地区居住着5亿人口的老龄化人口,随着人口老龄化,他们面临着与医疗设备故障和不良事件相关的更大风险。这是法规关注产品生命周期的主要原因之一,而不仅仅是进入市场。
4.欧洲医疗器械法规(MDR)涵盖哪些设备?
MDR将术语“医疗设备”定义为用于以下任何一项的“仪器,仪器,器具,软件,植入物,试剂,材料或其他物品”:
a、诊断,预防,监测,治疗或减轻疾病,残疾或伤害,但不用于残疾或伤害预防
b、调查,替换或修改解剖学,生理学或病理学过程
c、通过体外检查来自人体的样品提供数据
d、该定义涵盖了广泛的现有设备,但并非全部。MDR新指定了需要获得CE标志的某些类型的产品,包括用于清洁,消毒或消毒医疗设备的产品,以及用于控制和支持受孕的设备,无论是通过药理学,免疫学还是代谢方式。
e、请务必直接查阅法规,以确定您的医疗器械是否属于MDR。即便如此,根据您的设备分类,您可能不会遇到一些合规困难。根据MDR,I类设备不需要由第三方审核员(指定机构)进行QMS审核。
5. 欧洲医疗器械法规(MDR)附件XVI涵盖哪些设备?
新的MDR共包含16个附件部分,最引人注目的部分是附件XVI。本节要求MDR涵盖某些设备 - 以前可能不被视为医疗设备的设备。因此,一些公司首次受到医疗器械合规性规定的约束。
附件十六要求下列产品组符合MDR的要求:
a、隐形眼镜和眼睛内或眼睛上使用的其他产品(此处将包括滴眼液和化妆品隐形眼镜)
b、通过手术侵入手段将产品引入体内以修改解剖结构(硅乳房植入物现在符合条件)
c、用于面部或其他皮下填充物的产品和物质(肉毒杆菌注射)
d、用于吸脂,脂肪分解或脂肪成形术的设备
e、用于纹身和脱毛的高强度辐射设备
f、使用电流或磁流刺激大脑的设备
i、附件XVI填补了以前MDD法规中存在的许多空白,特别是将其自身强加于逃避监管的程序中使用的设备,因为它们是化妆品而非医疗性质。
6. 欧洲医疗器械法规(MDR)是否需要增强的设备可追溯性?
a是的,它会的。新的MDR包括唯一设备识别(UDI)的授权,旨在促进该地区销售的所有医疗设备的可追溯性。设备必须标有设备标识符(DI),产品的每个批次或生产系列都将标有生产标识符(PI)。
bMDR还为临床调查,产品注册和上市后监督引入了新的数据库。该EUDAMED数据库将用作:允许指定机构的多个数据库,医疗器械企业,消费者,监管者和其他利益相关者的系统来访问在欧洲销售的医疗器械的最新数据的一部分。
7. 欧洲医疗器械法规(MDR)将如何影响CE认证标志?
目前,新的MDR对获得CE标志没有显着影响。尽管最终文件于2017年5月发布,但该法规要到2020年5月才能生效,这使得医疗器械公司有足够的时间和机会实现合规。
截至2020年5月26日生效日期,医疗器械公司仍可获得公告机构的合规认证,这些证书自发布之日起五年内有效,可实现平稳过渡期。在生效日期之前合法投放市场的设备可以在MDR生效后五年内出售,但其合规证书将于2024年5月25日全部失效。
8.新的欧洲医疗器械法规(MDR)将如何影响我的质量管理体系?
一旦新的MDR于2020年5月生效,任何和所有以前的“指令”将不再存在或适用于质量体系。相反,他们将有一个新的名称,并带有更实质的意义 - 法规。简单的翻译是,这些变化现在将被视为法律 - 类似于FDA医疗器械法规。
制造商应遵循现在所谓的“一般义务”,以确保其质量管理体系(QMS)的合规性。这个全面的清单可以从第X条开始在新的MDR中找到。
如MDR指南中所述,制造商应建立,记录和实施质量体系,并在整个设备生命周期内保持其有效性。此外,还强调QMS 的管理功能,包括文档存储,上市后监控以及新设备和现有设备的风险评估所需的程序。
在QMS实施期间,制造商必须以质量手册和书面政策/程序的形式包括文档,包括:
1.质量目标。
2.组织业务。
3.用于设备控制,验证,验证和设备审查的程序/技术。
4.制造阶段的质量保证和验证程序。
5.相关测试和试验。
如果制造商计划实施新的质量体系,他们必须提交“与认证机构一起评估其质量体系的申请”(不包括I类设备),申请文件必须包括:
1.制造商目前的质量管理体系和用于确保充分性和有效性的程序。
2.制造商的PMS系统和临床评估计划,以及这两个程序的描述,以确保这些程序将保持最新。
2.必须记录用于满足QMS和MDR / IVDR规定的当前程序以及每个程序的描述。
3.到2020年,3年过渡期将结束。在5月26 日之后,制造商必须为计划在欧盟市场销售的每台设备实施所有新的QMS规定。
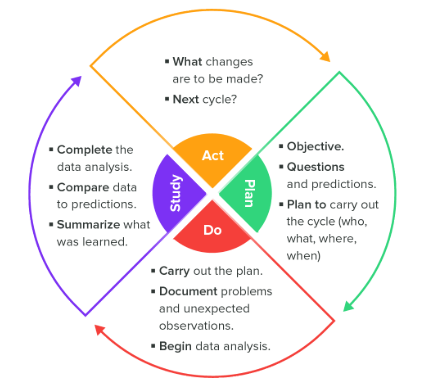
结论
欧洲MDR体现了未来十年医疗器械监管环境将如何变化。新发布的ISO 13485:2016和MDSAP计划已经在推动更高的标准化和更强的上市后监督要求,以及面向流程的风险管理和设备管理的生命周期方法。
尽管MDR的生效日期在未来很遥远,但今天的合规性承诺将使您的医疗公司在长期的行业繁荣中取得空前成功,并在满足全球有价值市场的监管合规目标方面取得空前成功。