tangxie520 第27页
-

欧盟REACH-REACH附录XVII条目解释性指南
欧盟委员会发布了一份解释性指南,以阐明打算涵盖的条款范围,并在REACH附件XVII的第72条下列出CMR(致癌,诱变或生殖毒性)物质的分析方法。除传统的服装和鞋类物品外,以下物品也可被视为在条目72的范围内,因为它们包含的纺织品在正常或合理可预见的使用条件下有望与人的皮肤接触,类似于服装:纺织品玩具(毛绒玩具,游戏垫)化装和化装;表带;家具和青少年产品的内饰;限制涵盖的CMR物质的可用分析方法如下:CMR物质[CAS号]重量浓度极限分析方法甲醛[50-00-0]<75毫克/公斤EN ISO 14184-1:...
-

EN 286 空气接收器:简单的压力容器指令
简单的压力容器是指打算在表压大于0.5 bar但小于或等于30 bar的条件下容纳空气或氮气并且不打算暴露于热的容器,必须批量生产,并且必须是具有总压力的焊接钢或铝结构,产品的体积不超过10,000bar.L。该指令根据存储的能量对船舶进行分类,以最大工作压力(以巴为单位)与其容量(以升为单位)(PS.V)的乘积表示,不同的规定适用于每种船舶,A类由PS.V大于5 0 bar.L的船只组成,并分为3个子类别。A类船舶的安全要求是满足指令附录1概述的基本安全要求,并在其上标明...
-

欧盟提议修订关于物质和混合物的CLP法规
欧盟提议修订关于物质和混合物的CLP法规欧盟已向WTO通报了其打算修改CLP法规的意图,要求在修订生效之日起18个月内对物质和混合物进行分类,标记和包装。2020年7月21日,世界贸易组织(WTO)宣布了欧盟(EU)的一项法规草案,旨在修订有关物质和混合物的分类,标签和包装的法规(EC)1272/2008(CLP法规) ,世贸组织第20-5008号文件所附的立法草案包含几项重要更改:1.在CLP法规“有害物质协调分类和标签清单”附件VI第3部分的表3中添加22个条目(“表3”);2.替换“表3”中的41个条目-其中...
-

欧盟对纺织品和鞋类产品的市场监督
实施《欧盟市场监管和产品合规性法规》(第(EU)2019/1020号法规)旨在防止将任何不合规产品投放到欧盟市场上并出售给欧盟消费者。2019年6月25日在《欧盟官方公报》(SafeGuardS 97/19)上发布的有关市场监控和产品合规性的(EU)2019/1020法规将于2021年7月16日适用,一些规定。它加强了非食品产品的市场监管并使之现代化,以保护公民免受不安全和不合格产品的侵害;因此,制造投放欧洲市场的所有产品都是为了确保安全并符合欧盟统一法规。2020年7月1日,欧洲化学品管理局(ECHA)宣布其检查...
-

门、窗户统一产品标准 EN 16034
什么是EN 16034?EN 16034是欧洲门,门和窗户的统一产品标准,该标准规定了对防火和防烟性能的要求。必须始终将其与EN 14351-1组合用于外部行人门套件或将EN 14351-2组合用于内部人行门套件(在欧盟官方杂志上发布并进行协调后)。自2016年推出以来,EN 16034标准已与国家法规保持一致并生效。该阶段称为共存阶段。共存阶段于2019年11月1 日结束,EN 16034自此生效;因此,从那时起,对于在市场上投放的产品,没有国家对外部门窗的批准是有效的,并且必须对该元素进行(CE-)标记以及性能...
-
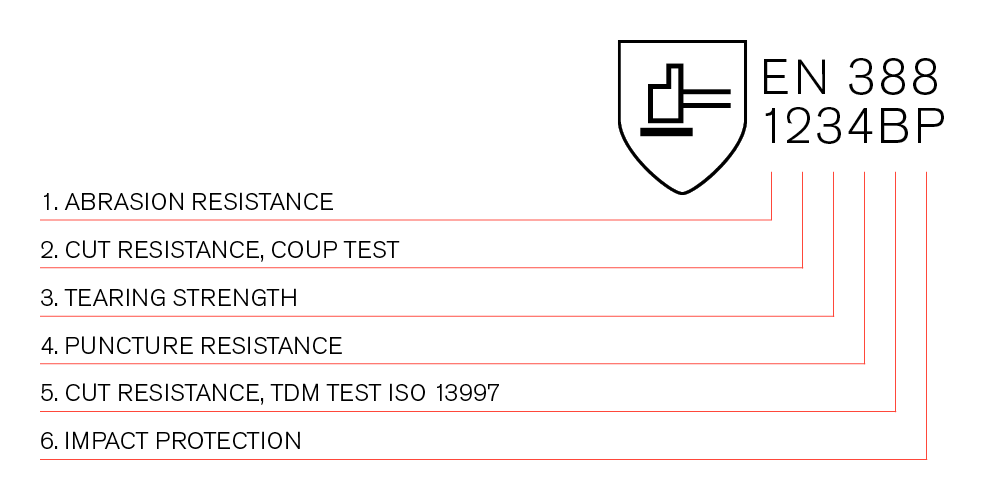
防机械风险防护手套EN 388:2016
防机械风险防护手套EN 388:2016根据该标准,测试了诸如耐磨性,耐切割性,抗撕裂强度,抗穿刺性和冲击防护等特性,结合象形图,将显示四个数字和一个或两个字母,这些迹象表明手套的性能。1.耐磨性在确定的压力下,用砂纸对材料进行磨损,EN 388:2016防护等级以1到4的比例表示,具体取决于材料上出现孔之前所需的匝数,数值越高,耐磨性越好。2.耐切割性,政变测试EN 388:2016切割保护已测试,将刀穿过手套材料,直到其穿过,保护级别由1到5之间的数字给出,其中5表示最高的割断保护,如果在此测试中材料使刀变钝,...
-

IEC/EN 62196_插头,插座,车辆连接器CE认证
IEC 62196的本部分适用于插头,插座,车辆连接器和带有标准配置的插脚和接触管的车辆进口,在本文中称为附件,它们的额定额定工作电压不超过480 V ac,50 Hz至60 Hz,并且额定电流不超过63 A三相或70 A单相,用于电动汽车的导电充电。这部分IEC 62196盖如在指定车辆供电的基本接口附件IEC 62196-1,和用于在传导充电系统中使用的用于在指定的电路IEC 61851-1:2010。IEC 62196 插头,插座,车辆连接器和车辆入口–电动汽车的导电??性充电是一系列国际标准,这些标准定义了...
-

EN 61851_电动汽车充电桩CE认证标准
电动汽车导电充电系统-第1部分:一般该标准还适用于由现场存储系统提供的电动汽车供电设备(例如,缓冲电池)。本标准涵盖的方面包括:1.电动汽车供电设备的特性和操作条件;2.电动汽车供电设备和电动汽车之间的连接规范;3.电动汽车电源设备的电气安全要求。EN 61851标准目的: IEC 61851的本部分适用于车载和非车载设备,用于以高达1000 V的标准交流电源电压(按照IEC 60038)和高达1500 V的直流电压为电动道路车辆充电连接到供电网络时,根据需要为车辆上的任何其...
-

自行车头盔安全EN 1078:2012 + A1:2012
自行车头盔安全EN 1078在欧盟,用于骑自行车的人和滑板手的头盔必须经过严格的测试和EC型式检验,由于骑自行车,溜冰和滑板等体育运动的性质,许多事故涉及速撞,因此,始终建议这些活动的参与者穿着防护服和头饰,以防止严重伤害。此类头部保护器的当前标准为EN 1078:2012 + A1:2012,请注意,在骑电动自行车时,这些头盔可能不合适。EN 1078包含许多一般要求,例如,材料必须安全无毒,即对穿戴者或与产品接触的其他任何人都不会造成危害。头盔的一个重要方面是,如果将其与洗护用品中常见的物质接触,则不得更改材料...
-

如何获得所需的机械CE认证资料?
为什么有必要做ce认证在欧洲市场上买卖机械或设备时,每家公司或实体都必须获得CE标志,通过机械CE认证流程,制造商可以声明其产品符合欧盟指令的所有健康和安全要求,欧盟机械占据了世界市场的三分之一;最大限度地减少机械事故是欧盟的一项基本政策,CE标志不是质量标志,它是制造商保证产品符合欧盟相关产品安全法的所有要求。机械CE认证是首次将产品投放市场或投入使用的人员的责任,负责方可以是:1.制造商(大多数情况下);2.制造商的授权代表;3.非CE标志产品的进口商进入欧洲;欧洲任何个人生产产品的用户;什么是机器指令2006...
